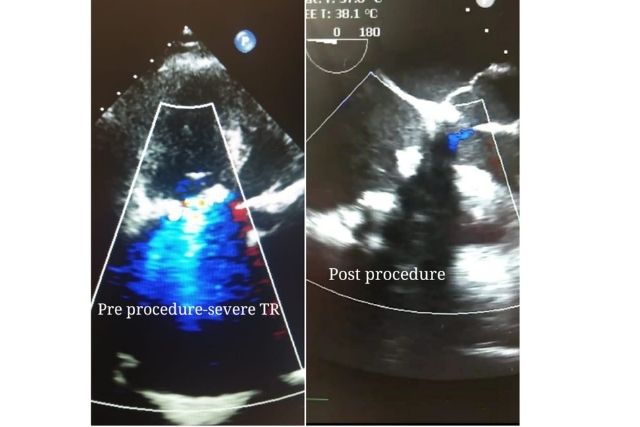
The testing and subsequent approval of a new medical device for commercialization is important for its safety, efficacy, and compliance with regulations. Medical devices can be quite simple such as a thermometer or advanced as a pacemaker and all of them go through the same series of steps before they are introduced into the market.
These steps are put in place so that the requirements for patient care, safety, and performance standards are met.
Conceptualization and Design
The process starts from a design and conceptualization stage. Targeting specific medical needs, engineers, medical practitioners, and people in charge of regulations work together to produce a prototype that is not only effective but also safe. During this stage, issues such as how effective and usable a device is, as well as its cost, are being considered while making sure that the device will be safe and effective.
Preclinical Testing
These studies attempt to evaluate the basic performance of said medical device for animals, or during laboratory studies—before the device is tested in human trials. This phase judges the integral principles of the device, and its biocompatibility along with potential risks of the device. Preclinical trials are a way to nip in the bud any possible concerns before clinical trials are conducted by use of the device.
Clinical Trials
When the preclinical evaluation of a particular device achieves satisfactory results, the next stage performed on human subjects (clinical trial) is begin.
This stage is subdivided into three phases:
- Phase 1 (The assessment of safety and associated side effects and complications): A small subset of healthy individuals (or at times patients) uses the device in controlled conditions. The main concern here is to determine the safety of the device and to document any associated complications or side effects.
- Phase 2 (The use of the device in clinical practice): Patients who suffer from the disease that is supposed to be cured by the device are involved in the trials. This stage determines the functional integrity of the device – for instance, whether it performs the anticipated therapeutic function.
- Phase 3 (Final improvements and confirmation of the efficacy and safety): A larger number of patients, who are not refractory to the tested product, is expected to be used to better verify the safety and the efficacy of the device. If the outcome is satisfactory, then the data can be put together for regulatory approval.
The Regulators’ Provision
When the clinical trials are completed successfully, the manufacturers start seeking a license for the device. In the USA this is done by the Food and Drug Administration (FDA) while in Europe it is the responsibility of the European Medicines Agency (EMA). The data of the device, including other materials like preclinical and clinical results, is examined to ascertain whether the authority’s standards in relation to safety, effectiveness and quality are being met.
If a device is likely to present a high risk to the health of patients, such as a pacemaker or a surgical implant, the FDA will most likely need one form of Premarket Approval (PMA) which is a more extensive review process. In the moderate level of risk involved devices, the manufacturers could submit application for 510(k) which precludes a demonstration of the new device to an extent that it is safe and effective as the device that has already been approved.
Post-Market Monitoring
When a medical device has been approved and put on the market, the postmarket phase of the study starts. This stage involves active follow up of the device in the market with the intention of determining any adverse effects or risks resulting from the use of the device that was not noted in the clinical trial. Specific issues are available and fall within the precautions of regulatory bodies, manufacturers, and healthcare providers to report any problems, and if some of the problems start occurring, the device may be re-evaluated.
Developments and Enhance
In most cases, the device undergoes further development after its entry onto the market. Healthcare providers and patients can request enhancements, upgrades, or patches that assist the device perform better or mitigate new threats. Ongoing improvements guarantee safety and effectiveness of medical devices over time.
Conclusion
The activity of evaluating and licensing new medical devices is complex, systematic and time-consuming procedure that assures devices can be used by patients without any harm reaching appropriate level of safety and efficiency. Everything from the concept through design verification and validation, clinical trials, regulation and marketing and post-marketing vigilance has its definite contribution into the security of the medical devices and their relevance to the task at hand if other members of the medical devices family are advanced. As the technology regarding medical devices advances, so too must the process to test and approve those medical devices to ensure that public health is safeguarded.