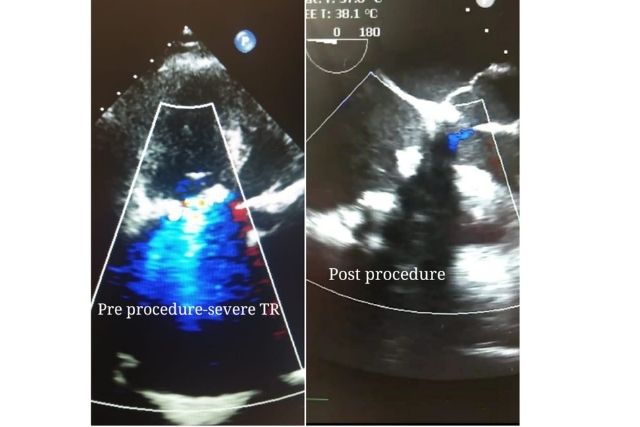
There is no denying that regulations reduce risks in medical devices’ design and development, as well as help to deliver safe and effective marketed products to patients. Medical devices are very much within the purview of regulation as they potentially can affect public health. Regulatory marketing frameworks govern the entire life cycle of the product, right from inception to post marketing surveillance in an optimized manner with creativity and safety.
Regulations in Medical Devices: Ensuring Safety and Innovation
Defining the Regulatory Framework
As with any other product on the marketplace, there are always rules associated with its development and exchange. These regulations are set by the governmental and regulated organizations in charge of assuring the safety & efficacy along with the quality of the medical product. These regulations change from one region to the next one… but the objectives of the regulations are quite the same: protect the health of the patients and stimulate creativity.
As far as the regulation of medical devices is concerned, its primary body in the USA is the FDA, whereas in Europe, it’s the role of the EMA and the NBs to regulate compliance with MDR. Other countries like Japan and Canada have different regulatory authorities who also set procedures for the endorsement of medical devices.
Steering the Stages of Artistic Design and Developmental work
Regulations intervene even during the initial stages of device building. The authorities provide definitions of a medical device and determine the optimal criteria such as QMS, design control, and risk management processes. Such guidelines ensure that the concerns of the manufacturers on device development are done stepwise to avoid potential risk during development and throughout the devices control plan.
In this regard for instance the FDA in its directive provides that Good Manufacturing Practices (GMP) be adhered to in any device production in this case, practices that ensure consistency in quality among produced devices. Similarly, ISO 13485 which the medical devices quality management system international standard provides a framework for the manufacturers during the device design to ensure it is within the legal requirements.
Risk Classification and Evaluation
One of the most apparent repercussions regulations have over the medical device development portion is the classification of risk. Based on certain considerations imposed by the medicine or device they are supposed to take onto the market; there are devices with low-risk like band-aids while on the other side there are high-risk devices like pacemakers.
The risk classification is determined by the relevant authorities and depending on that criteria devices are looked into according to that class and the strictness for that class. For instance,
- Class I (Low Risk): Such Devices Like tongue depressors or handheld surgical instruments and having general controls, such as label requirements, and general level of good manufacturing practices.
- Class II (Moderate Risk): A device like an infusion pump or any diagnostic equipment that requires premarketing submission to establish its substantial equivalence to xenogenetic devices is approved under this classification, such as FDA’s 510(k) clearance.
- Class III (High Risk): Heart valves and implanted defibrillators are such devices that require rigorous PMA (sect 515(c)) to be able to show that it is reasonably safe and effective before marketing.
Regulations make sure that devices are subjected to the right level of testing, clinical, and data collection, depending on the classification of the device.
Clinical Trials and Evidence Based Testing
Evidence based testing is obligatory to such devices as medical devices are always expected to be safe and effective. This may include the need for a clinical study for high-risk characteristics or provide modest device performance evaluations for lower-class devices. With clinical trials, the FDA has also mandated the forth validation of the devices for safety and efficacy before marketing for more advanced devices.
Regulations further assist in the determination and the provision of the evidences that are required to support the approval of a device which may include the biocompatibility data the performance data and the device longevity data. Such agencies might require the devices to be monitored on a long term basis for any of the risks that might have escaped all of the pre marketing tests.
Regulatory Approval and Market Authorization
Finally, after the conclusive success of the trials and evaluations, the manufacturers go ahead and apply for regulatory approval, which is also though to take some time area.
The level at which the application is made differs depending on the area:
- FDA (U.S.): In most cases, for Class II devices, manufacturers are required to file 510(k) notification in order to show that a new device developed is substantially equivalent to another device that has already been legally marketed. For Class III devices however, a more detailed process known as Premarket Approval (PMA) is mandatory for the manufacturers.
- European Union (EU): On the other hand, in Europe, Class II and III devices have to go through a conformity assessment process by the EU notified body, which confirms that all essential requirements under the Medical Device Regulation (MDR) were met.
After the application has been accepted, the device is granted a marketing authorization order. However, that is not the end of the regulatory procedure.
Post-Market Surveillance and Compliance
Regulatory provisions go further than the marketing stage where manufacturers are tasked with responsibility of ensuring that their marketed devices are safe and performing the intended purpose.
This includes:
- Post-market surveillance: Manufacturers are required to notify of the adverse events that occur at the clinical setting when the device is utilized and regarding any incident of device failure.
- Quality control audits: Regulatory institutions may carry out inspections almost on-site such as; post-marketing studies, surveillance after licensing.
The idea behind post-market regulation is that all the devices undergo regular checks and any alterations or recall that are necessary should be performed without delay throughout the course of the device lifecycle.
Incorporating New Technology
Every time new medical technology is introduced, it is the role of regulatory bodies to ensure that the regulatory frameworks are constantly updated. Regulators are expected to remain relevant due to the advent of new technologies such as digital health, nuts and bolts of AI, and the advancement of precision medicine. In response, the FDA and EMA agencies have both been developing newer guidelines in order to suit these technologies so that newer technologies can go through the same processes as older medical technologies.
For example, under the US FDA there is a Digital Health Innovation Action Plan, which allows for a quicker process in the approval of software developed medical devices and mobile apps for health, serving as a target for developers of new age technologies.
Conclusion
Difficulties do exist in the process of development of medical devices with numerous aspects revolving around regulations which ensure that manufacturers adhere to guidelines from the idea of a device to its commercialization. These regulations, while different in strictness and form across various regions of the globe, all aim at protecting the patients. Harm can be caused only if regulatory authorities are incapable of finding a fine line between safeness planning and flawlessness thus artificial limits to forecasting the discovery of a new medical device can be put on.